学术动态
浙江大学周青课题组《Nature》发文报道RIPK1切割位点变异可导致自身炎症性疾病
作者:周青 来源自:中国免疫学会 点击数:65768 发布时间:2020-06-12
自身炎症性疾病是由先天免疫系统异常激活引起的一组罕见疾病,其特征为无诱因的发烧和炎症,可能损害重要器官。例如:家族性地中海热(FMF),IL-1受体拮抗剂缺乏症(DIRA)等。由于炎症是周期性反复发生的,这些疾病也被称为“周期性发热综合征”。 自身炎症性疾病是由于调节先天免疫系统的基因发生突变导致的,这些遗传变异可以从父母传给孩子,导致家庭中出现多例相似的患者。医学遗传学的发展使科学家能够鉴定出导致不同自身炎症性疾病的遗传变异,为患者的临床治疗和罕见病的基因检测提供宝贵的数据基础。
RIPK1(Receptor-Interacting serine/threonine-Protein Kinase-1)是受体相互作用蛋白(RIP)激酶家族的一员,参与了决定细胞“生死存亡”的多种重要信号通路,其激酶活性在RIPK1依赖的细胞凋亡和细胞程序性坏死进程中发挥重要的调节作用。RIPK1蛋白的中间结构域(intermediate domain)中含有caspase-8的切割位点(Asp324)。以往的研究发现,小鼠RIPK1该位点突变会引起胚胎致死,主要是由于增多的细胞凋亡和程序性坏死。然而,人类RIPK1该切割位点变异对这些重要信号通路及人类健康的影响未有报道。
2020年1月2日,浙江大学生命科学研究院周青课题组、哈佛大学医学院袁钧瑛课题组和复旦大学附属儿科医院王晓川课题组联合在《Nature》杂志上发表研究论文“A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1”,报道了人类RIPK1切割位点变异会阻止caspase-8蛋白酶对其切割,导致细胞凋亡、细胞坏死和炎症信号通路的过度激活,从而引起严重的自身炎症性疾病。利用揭示的发病分子机制指导临床治疗,并极大的缓解了病人的临床症状。
周青课题组从来自世界各地的、未能明确诊断的自身炎症疾病病例中,通过全外显子测序,鉴定到了两个分别来自中国和加拿大的家系,患者的RIPK1蛋白均有D324氨基酸新发突变。病人表现出典型的自身炎症疾病表型,主要包括周期性发热、淋巴结肿大、肝脾肿大等(图1)。
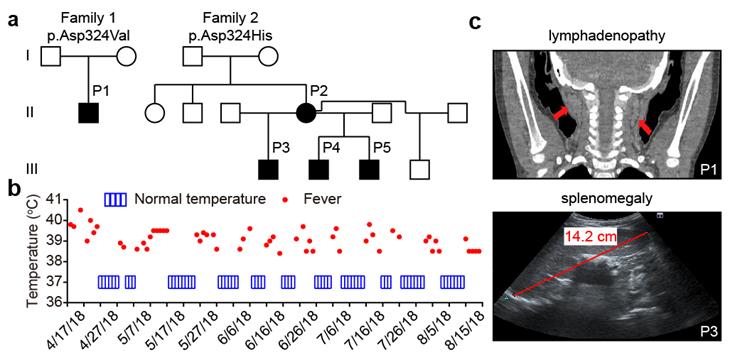
图1. 两个携带RIPK1蛋白突变的家系遗传及表型
为了确认外显子测序对突变鉴定的准确性以及明确病人的炎症指标,作者通过Sanger测序验证所有病人均确实携带RIPK1杂合突变,并取病人全血分别分离血清和外周血单个核细胞(PBMCs)以检测炎症信号。血清细胞因子检测发现病人血清中促炎细胞因子IL-6、TNF和抗炎细胞因子IL-10等水平显著升高;PBMCs的单细胞转录组学分析发现病人的单核细胞比例提高,并且NF-kB和type-I IFN炎症信号通路基因转录水平显著上升。
鉴于RIPK1蛋白在细胞死亡和炎症信号通路中的重要作用,作者接下来分别对来自病人和健康对照的PBMCs进行程序性死亡诱导,结果发现,携带RIPK1杂合变异的病人细胞对TNF诱导的细胞凋亡和细胞程序性坏死更加敏感,并且可以诱导产生显著上调的IL-6、TNF等炎症因子,这些变化可以通过添加RIPK1激酶抑制剂necrostatin-1s (Nec-1s) 缓解,说明其都依赖于上调的RIPK1激酶活性。Cyclophilin A 的释放在细胞坏死后会增加,已作为一种生化标志物应用于多种疾病的诊断。研究人员发现,携带RIPK1变异的病人尿液中可以检测到cyclophilin A 水平相比健康对照显著升高。这些结果分别从体外和体内证明了RIPK1 D324位点的变异促进了RIPK1的激活,进而导致细胞死亡的增加和自身炎症的发生。
为了进一步弄清楚病人发病的分子机制,作者在Ripk1-knockout MEFs中分别过表达WT和D325突变的RIPK1,结果与PBMCs实验结果一致,D325位点突变的RIPK1在TNF诱导的细胞凋亡和细胞坏死过程中激活显著增加,进而引起细胞死亡水平的显著上调。而细胞坏死缺陷的RIPK1 D325突变的knock in小鼠(Ripk3-/-Ripk1D325A/D325A)实验同样表现出对TNF诱导的细胞凋亡更加敏感,说明过度激活的RIPK1对细胞凋亡和细胞坏死的促进是相互独立,互不依赖的。炎症因子检测的实验数据也证明在TNF的刺激下,RIPK1 D325突变的MEFs细胞中IL6、CXCL2和TNF转录水平显著上调。
通过对病人的血清、PBMCs细胞和RIPK1 D325突变的MEFs细胞的细胞炎症因子实验结果分析发现,RIPK1过度激活显著提高了IL6水平。因此,作者建议对病人采用tocilizumab (anti-IL-6R)进行临床治疗用,相比之前采取的治疗方式,tocilizumab极大地缓解了病人的炎症症状。
实验中一个有趣的发现是,与病人PBMCs相反,病人的皮肤成纤维细胞 (fibroblasts) 表现出对TNF、LPS诱导的细胞坏死的抵抗,而且对Erastin、RSL3等诱导的铁死亡也有抵抗作用。进一步研究发现,病人成纤维细胞RIPK1、TNFR1等蛋白表达水平明显下调,细胞内的还原性谷胱甘肽(GSH)含量高,活性氧(ROS)含量低,这些变化部分解释了成纤维细胞对不同死亡形式的抵抗。这一现象提示病人成纤维细胞为应对RIPK1变异导致的对多种刺激的高敏感性发展出多种补偿机制来维持机体稳态。
本研究首次发现人类RIPK1非切割变异导致自身炎症性疾病。解析其发病分子机制:不能切割的RIPK1可以促进其激酶活性,导致细胞凋亡、细胞程序性坏死增加、炎症因子上调,为临床提供更加合理、科学的治疗建议;同时还发现病人不同种类细胞响应外来刺激的异质性,丰富了RIPK1在调节不同种类细胞死亡中的作用。
浙江大学生命科学研究院周青研究员、哈佛大学医学院袁钧瑛教授、复旦大学附属儿科医院王晓川教授和俞晓敏研究员为本文通讯作者。周青课题组博士研究生陶攀峰、博士研究生王俊和王诗豪博士,袁钧英课题组吴哲铭博士、李婉津博士和复旦大学附属儿科医院孙金峤教授为本文共同第一作者。
原文链接:https://www.nature.com/articles/s41586-019-1830-y
RIPK1(Receptor-Interacting serine/threonine-Protein Kinase-1)是受体相互作用蛋白(RIP)激酶家族的一员,参与了决定细胞“生死存亡”的多种重要信号通路,其激酶活性在RIPK1依赖的细胞凋亡和细胞程序性坏死进程中发挥重要的调节作用。RIPK1蛋白的中间结构域(intermediate domain)中含有caspase-8的切割位点(Asp324)。以往的研究发现,小鼠RIPK1该位点突变会引起胚胎致死,主要是由于增多的细胞凋亡和程序性坏死。然而,人类RIPK1该切割位点变异对这些重要信号通路及人类健康的影响未有报道。
2020年1月2日,浙江大学生命科学研究院周青课题组、哈佛大学医学院袁钧瑛课题组和复旦大学附属儿科医院王晓川课题组联合在《Nature》杂志上发表研究论文“A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1”,报道了人类RIPK1切割位点变异会阻止caspase-8蛋白酶对其切割,导致细胞凋亡、细胞坏死和炎症信号通路的过度激活,从而引起严重的自身炎症性疾病。利用揭示的发病分子机制指导临床治疗,并极大的缓解了病人的临床症状。
周青课题组从来自世界各地的、未能明确诊断的自身炎症疾病病例中,通过全外显子测序,鉴定到了两个分别来自中国和加拿大的家系,患者的RIPK1蛋白均有D324氨基酸新发突变。病人表现出典型的自身炎症疾病表型,主要包括周期性发热、淋巴结肿大、肝脾肿大等(图1)。
图1. 两个携带RIPK1蛋白突变的家系遗传及表型
为了确认外显子测序对突变鉴定的准确性以及明确病人的炎症指标,作者通过Sanger测序验证所有病人均确实携带RIPK1杂合突变,并取病人全血分别分离血清和外周血单个核细胞(PBMCs)以检测炎症信号。血清细胞因子检测发现病人血清中促炎细胞因子IL-6、TNF和抗炎细胞因子IL-10等水平显著升高;PBMCs的单细胞转录组学分析发现病人的单核细胞比例提高,并且NF-kB和type-I IFN炎症信号通路基因转录水平显著上升。
鉴于RIPK1蛋白在细胞死亡和炎症信号通路中的重要作用,作者接下来分别对来自病人和健康对照的PBMCs进行程序性死亡诱导,结果发现,携带RIPK1杂合变异的病人细胞对TNF诱导的细胞凋亡和细胞程序性坏死更加敏感,并且可以诱导产生显著上调的IL-6、TNF等炎症因子,这些变化可以通过添加RIPK1激酶抑制剂necrostatin-1s (Nec-1s) 缓解,说明其都依赖于上调的RIPK1激酶活性。Cyclophilin A 的释放在细胞坏死后会增加,已作为一种生化标志物应用于多种疾病的诊断。研究人员发现,携带RIPK1变异的病人尿液中可以检测到cyclophilin A 水平相比健康对照显著升高。这些结果分别从体外和体内证明了RIPK1 D324位点的变异促进了RIPK1的激活,进而导致细胞死亡的增加和自身炎症的发生。
为了进一步弄清楚病人发病的分子机制,作者在Ripk1-knockout MEFs中分别过表达WT和D325突变的RIPK1,结果与PBMCs实验结果一致,D325位点突变的RIPK1在TNF诱导的细胞凋亡和细胞坏死过程中激活显著增加,进而引起细胞死亡水平的显著上调。而细胞坏死缺陷的RIPK1 D325突变的knock in小鼠(Ripk3-/-Ripk1D325A/D325A)实验同样表现出对TNF诱导的细胞凋亡更加敏感,说明过度激活的RIPK1对细胞凋亡和细胞坏死的促进是相互独立,互不依赖的。炎症因子检测的实验数据也证明在TNF的刺激下,RIPK1 D325突变的MEFs细胞中IL6、CXCL2和TNF转录水平显著上调。
通过对病人的血清、PBMCs细胞和RIPK1 D325突变的MEFs细胞的细胞炎症因子实验结果分析发现,RIPK1过度激活显著提高了IL6水平。因此,作者建议对病人采用tocilizumab (anti-IL-6R)进行临床治疗用,相比之前采取的治疗方式,tocilizumab极大地缓解了病人的炎症症状。
实验中一个有趣的发现是,与病人PBMCs相反,病人的皮肤成纤维细胞 (fibroblasts) 表现出对TNF、LPS诱导的细胞坏死的抵抗,而且对Erastin、RSL3等诱导的铁死亡也有抵抗作用。进一步研究发现,病人成纤维细胞RIPK1、TNFR1等蛋白表达水平明显下调,细胞内的还原性谷胱甘肽(GSH)含量高,活性氧(ROS)含量低,这些变化部分解释了成纤维细胞对不同死亡形式的抵抗。这一现象提示病人成纤维细胞为应对RIPK1变异导致的对多种刺激的高敏感性发展出多种补偿机制来维持机体稳态。
本研究首次发现人类RIPK1非切割变异导致自身炎症性疾病。解析其发病分子机制:不能切割的RIPK1可以促进其激酶活性,导致细胞凋亡、细胞程序性坏死增加、炎症因子上调,为临床提供更加合理、科学的治疗建议;同时还发现病人不同种类细胞响应外来刺激的异质性,丰富了RIPK1在调节不同种类细胞死亡中的作用。
浙江大学生命科学研究院周青研究员、哈佛大学医学院袁钧瑛教授、复旦大学附属儿科医院王晓川教授和俞晓敏研究员为本文通讯作者。周青课题组博士研究生陶攀峰、博士研究生王俊和王诗豪博士,袁钧英课题组吴哲铭博士、李婉津博士和复旦大学附属儿科医院孙金峤教授为本文共同第一作者。
原文链接:https://www.nature.com/articles/s41586-019-1830-y