
Prof. Qiang Xu’s group reports how an autonomous activation of interleukin-17 receptor signaling promotes autoimmune disease progression
Source:Qiang Xu
2023-10-24
Interleukin-17 (IL-17) is a key pro-inflammatory cytokine produced mainly by Th17 cells. It is notably upregulated in patients with various autoimmune diseases including psoriasis, rheumatoid arthritis (RA), and multiple sclerosis (MS), and is closely associated with the development and progression of these diseases. Currently, directly targeting IL-17A, IL-17F and IL-17RA, or inhibiting IL-17 production are the mainstay clinical treatment for IL-17-related autoimmune diseases. However, except for a few diseases such as psoriasis, several clinical trials of secukinumab and brodalumab in IL-17-related diseases, including RA, MS and inflammatory bowel disease, have been terminated because it had no or limited clinical effect. The reasons for these contradictory outcomes, which contradict the underlying pathogenesis, remain unclear. This not only greatly limits the prospects of anti-IL-17 therapy in related diseases but also suggests the existence of unknown molecular players within the IL-17 signaling pathway.
On September 12, 2023, Qiang Xu’s group of Nanjing University published a research paper entitled “An autonomous activation of interleukin-17 receptor signaling sustains inflammation and promotes disease progression” in Immunity. It reveals that increased SHP2 interacts with Act1 to induce IL-17-independent IL-17R signaling, which leads to persistent inflammation and nullifies anti-IL-17 efficacy.
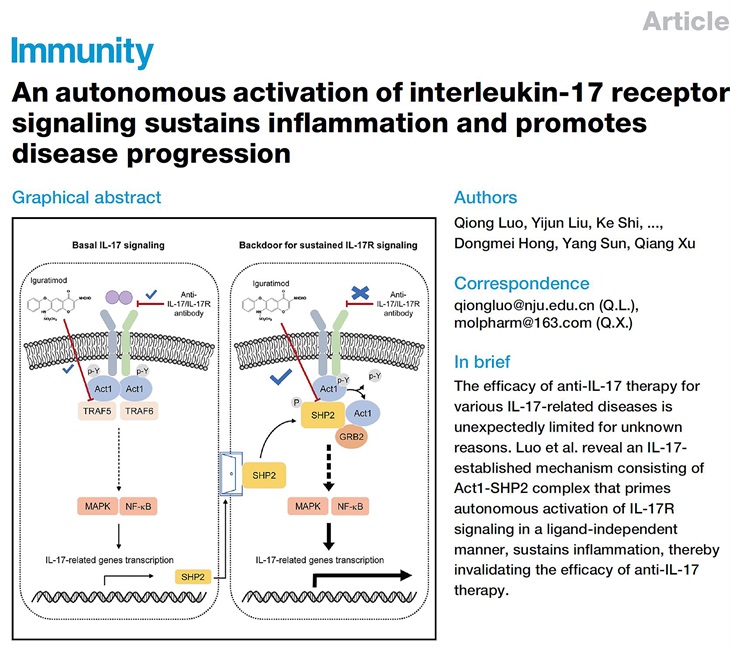
In this work, the researchers first found that the phosphatase SHP2 is highly expressed in lesional tissues of various autoimmune diseases, related animal models, and IL-17A-stimulated tissue cells. SHP2 overexpression alone (without IL-17A) increased CXCL1, CXCL2 and IL-6 production at both mRNA and protein levels. Although anti-IL-17A antibodies GR1501 and Cosentyx greatly inhibited the production of pro-inflammatory molecules induced by IL-17A, their inhibitory effects were completely abrogated upon SHP2 overexpression. In addition, when the administration started from day 31 (clinical score reached 3), the inhibitory effect of anti-IL-17A antibody on collagen-induced arthritis was no longer observed, and at this time, the expression level of SHP2 in the joint tissue is higher. Furthermore, SHP2 deficiency in astrocytes paralyses IL-17 signaling and attenuates EAE development. The researchers next found that SHP2 interacted with Act1, an adapter and essential component of IL-17 signaling. SHP2 competed with TRAF5 for Act1-binding and enhanced inflammatory signaling transduction. Mechanistically, SHP2 utilized its phosphatase activity to dephosphorylate Act1 and promoted both IL-17-induced and IL-17-independent activation of IL-17R signaling. The researchers further found that iguratimod blocked Act1-SHP2 interaction via occupying the TRAF domain of Act1, thus inhibiting both basal IL-17 signaling and IL-17-independent IL-17R signaling through blocking Act1 interactions with TRAF5 and SHP2 respectively. In addition, treatment with iguratimod from day 31 (clinical score reached 3) still showed a significant improvement in CIA, which was quite distinct from the anti-IL17 antibodies.
In conclusion, this study reveals an IL-17-established mechanism consisting of Act1-SHP2 complex that primes autonomous activation of IL-17R signaling in a ligand-independent manner, sustains inflammation, thereby invalidating the efficacy of anti-IL-17 therapy. They also provides mechanistic insights that may apply to various autoimmune diseases, as a similar backdoor mechanism may also exist in other inflammatory signal pathways, which may help pave an additional approach to the chronic diseases.
Dr. Qiong Luo and Dr. Qiang Xu of Nanjing University are co-corresponding authors of this paper. The work was funded by the National Natural Science Foundation of China and etc.
Links: https://www.cell.com/immunity/fulltext/S1074-7613(23)00269-8
On September 12, 2023, Qiang Xu’s group of Nanjing University published a research paper entitled “An autonomous activation of interleukin-17 receptor signaling sustains inflammation and promotes disease progression” in Immunity. It reveals that increased SHP2 interacts with Act1 to induce IL-17-independent IL-17R signaling, which leads to persistent inflammation and nullifies anti-IL-17 efficacy.
In this work, the researchers first found that the phosphatase SHP2 is highly expressed in lesional tissues of various autoimmune diseases, related animal models, and IL-17A-stimulated tissue cells. SHP2 overexpression alone (without IL-17A) increased CXCL1, CXCL2 and IL-6 production at both mRNA and protein levels. Although anti-IL-17A antibodies GR1501 and Cosentyx greatly inhibited the production of pro-inflammatory molecules induced by IL-17A, their inhibitory effects were completely abrogated upon SHP2 overexpression. In addition, when the administration started from day 31 (clinical score reached 3), the inhibitory effect of anti-IL-17A antibody on collagen-induced arthritis was no longer observed, and at this time, the expression level of SHP2 in the joint tissue is higher. Furthermore, SHP2 deficiency in astrocytes paralyses IL-17 signaling and attenuates EAE development. The researchers next found that SHP2 interacted with Act1, an adapter and essential component of IL-17 signaling. SHP2 competed with TRAF5 for Act1-binding and enhanced inflammatory signaling transduction. Mechanistically, SHP2 utilized its phosphatase activity to dephosphorylate Act1 and promoted both IL-17-induced and IL-17-independent activation of IL-17R signaling. The researchers further found that iguratimod blocked Act1-SHP2 interaction via occupying the TRAF domain of Act1, thus inhibiting both basal IL-17 signaling and IL-17-independent IL-17R signaling through blocking Act1 interactions with TRAF5 and SHP2 respectively. In addition, treatment with iguratimod from day 31 (clinical score reached 3) still showed a significant improvement in CIA, which was quite distinct from the anti-IL17 antibodies.
In conclusion, this study reveals an IL-17-established mechanism consisting of Act1-SHP2 complex that primes autonomous activation of IL-17R signaling in a ligand-independent manner, sustains inflammation, thereby invalidating the efficacy of anti-IL-17 therapy. They also provides mechanistic insights that may apply to various autoimmune diseases, as a similar backdoor mechanism may also exist in other inflammatory signal pathways, which may help pave an additional approach to the chronic diseases.
Dr. Qiong Luo and Dr. Qiang Xu of Nanjing University are co-corresponding authors of this paper. The work was funded by the National Natural Science Foundation of China and etc.
Links: https://www.cell.com/immunity/fulltext/S1074-7613(23)00269-8
