学术动态
陈玮团队《Nat Immunol》揭示E3泛素连接酶RAD18可终止IRF3驱动的IFNB1转录
作者:陈玮 来源自:中国免疫学会 点击数:11171 发布时间:2025-09-24
2025年8月29日,浙江大学医学院免疫学研究所陈玮团队联合浙江大学附属第二医院眼科中心韩伟团队在《Nature Immunology》杂志上发表题为“E3 ligase RAD18 targets phosphorylated IRF3 to terminate IFNB1 transcription”的研究论文。该研究突破性揭示E3泛素连接酶RAD18在抗病毒先天免疫中的“分子刹车”功能。其作用机制表现为:RAD18特异性识别磷酸化活化状态的IRF3(p-IRF3),通过K63型泛素化修饰促使p-IRF3从IFNB1启动子解离,并触发自噬-溶酶体途径降解。这一精密调控机制既有效抑制IFNβ的过度表达,又为RNA病毒感染(如流感病毒)和自身免疫性疾病(如系统性红斑狼疮)的靶向治疗提供了全新干预策略。
一个多世纪来RNA病毒大流行已成为人类健康的重大威胁, I型干扰素(IFN-Ⅰ)是一类具有抗病毒和抗肿瘤活性的细胞因子,作为机体免疫系统的重要组成部分,以其独特的抗病毒、抗细胞分裂以及免疫调节功能,发挥着举足轻重的作用。但其过度表达会导致多种病理生理改变,可能会导致免疫调节失衡造成组织病理损伤以及自身免疫性疾病(如系统性红斑狼疮)。合理控制I型干扰素的表达具有重要的生物学意义。
I型感染素的产生始于免疫细胞表面的模式识别受体(PRRs)对病毒等的病原体相关分子模式(PAMPs)的识别,通过TLR3/4-TRIF、RIG-I-MAVS和cGAS-STING信号转导通路触发核心转录因子IRF3的磷酸化,进而驱动I型干扰素的转录。值得关注的是,在高剂量的RNA病毒感染情况下,磷酸化IRF3蛋白(p-IRF3)在驱动I型干扰素转录之后,发生完全降解。这个现象表明机体通过促进p-IRF3降解从而避免过量IFNs产生以及不可控的炎症反应发生。然而,目前关于介导p-IRF3完全降解的关键机制并不清楚。研究人员首先通过质谱检测发现在细胞受到病毒刺激后,E3泛素连接酶RAD18与IRF3的结合大大增加,且能促进p-IRF3发生强烈的泛素化,而该泛素化与IRF3的自噬途径降解相关。进一步的研究发现RAD18只结合二聚体 p-IRF3,且二聚体 p-IRF3与DNA链的结合是其被RAD18蛋白识别的前置条件,这提示RAD18可能参与p-IRF3转录活性的调控。当细胞RAD18敲除后,p-IRF3蛋白的降解被抑制且滞留在细胞核内,同时细胞IFNB1的mRNA持续高表达,对病毒(包括SARS-CoV-2)的感染表现出更强的防御反应。机制研究发现E3泛素连接酶RAD18通过其独特的分子识别机制,特异性结合IFNB1启动子上的磷酸化IRF3,并催化形成K63连接的多泛素链修饰(靶向人IRF3的Lys193位点,对应小鼠IRF3的Lys188位点),这种泛素化修饰导致p-IRF3二聚体从转录复合物中解离,并触发其核质转运过程,从而终止IFNB1的转录激活。而其他与IRF3相关E3泛素连接酶(包括TRIM21、TRIM26、RBCK1和UBE3C等)均不具备直接调控p-IRF3转录活性的功能,突显了RAD18在该通路中的特异性调控地位。
研究人员通过Knock-in技术构建了IRF3-K188R(赖氨酸突变为精氨酸)基因突变小鼠,实验结果显示突变小鼠的巨噬细胞在应对病毒感染后,表现出更强的I型IFN反应。实验性变态反应性脑脊髓炎(EAE)造模结果也显示,突变小鼠因产生了更多的IFNβ,其脊髓炎症细胞浸润程度和脊髓脱髓鞘程度均较轻。研究人员还构建了Rad18fl/fl Lysm-cre 小鼠,体内病毒感染实验结果表明Rad18fl/fl Lysm-cre 小鼠在感染水泡性口炎病毒后的生存期更久,体内脏器和血清中的IFNβ因子表达更高。此外,研究人员的临床病理样本分析发现RAD18在系统性红斑狼疮(SLE)病人的外周血单核细胞中的表达较低,在活动期SLE患者中,RAD18蛋白水平分别与IRF3磷酸化水平和IFNB1/IFNA mRNA水平呈负相关,表明RAD18在SLE中对p-IRF3驱动的IFN表达具有负调控作用。这些研究内容为RAD18作为自身免疫性疾病的潜在治疗靶点提供了依据。
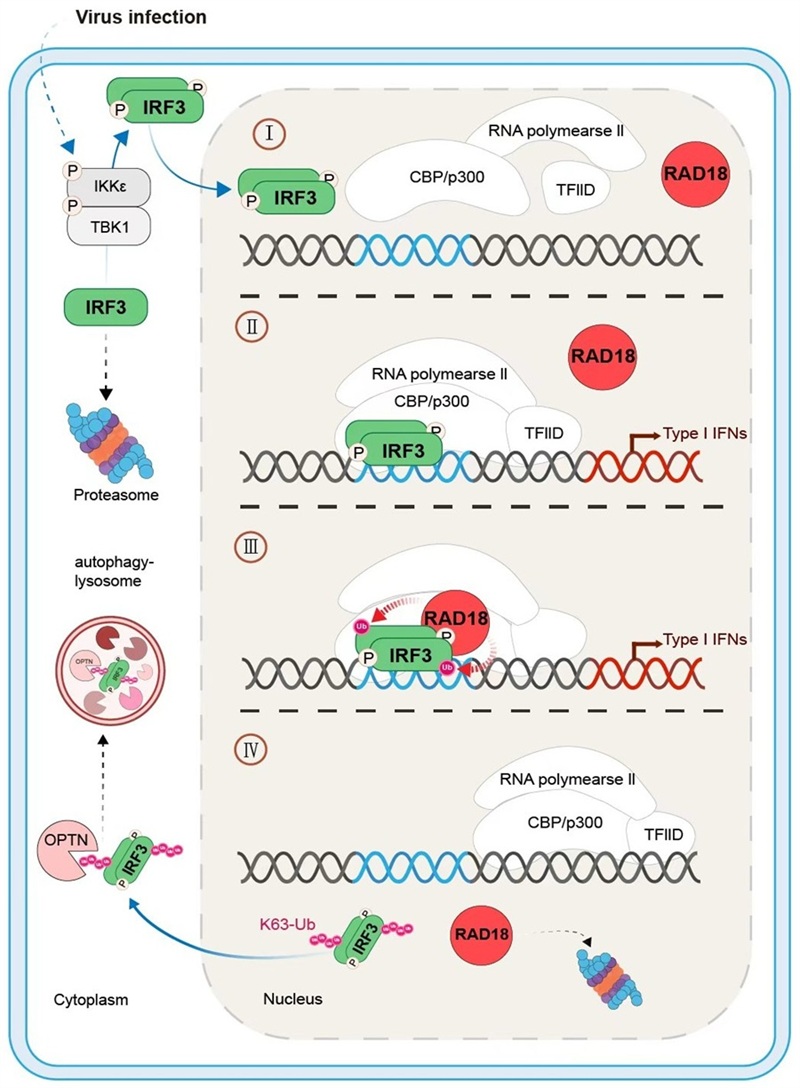
该研究详细阐明RAD18作为E3泛素连接酶在p-IRF3自噬降解通路中的核心调控作用。其分子机制表现为:RAD18特异性识别并结合IFNB启动子区域的p-IRF3二聚体,通过泛素化修饰终止p-IRF3的转录活性,从而精准调控IFNB1基因的表达。这一发现揭示了RAD18在IRF3信号通路中作为“制动器”的关键功能。
浙江大学医学院基础医学院的陈玮和浙大二院眼科中心的韩伟教授为该论文的通信作者。浙江大学医学院博士生蔡依廷、郑家琦博士、赵琳琳硕士和王雪硕士为本文共同第一作者。该研究得到国家自然科学基金和浙江省自然科学基金资助,并得到浙江大学免疫学研究所、免疫与炎症全国重点实验室和浙江大学医学院公共技术平台的帮助和支持。
原文链接:https://www.nature.com/articles/s41590-025-02256-x
一个多世纪来RNA病毒大流行已成为人类健康的重大威胁, I型干扰素(IFN-Ⅰ)是一类具有抗病毒和抗肿瘤活性的细胞因子,作为机体免疫系统的重要组成部分,以其独特的抗病毒、抗细胞分裂以及免疫调节功能,发挥着举足轻重的作用。但其过度表达会导致多种病理生理改变,可能会导致免疫调节失衡造成组织病理损伤以及自身免疫性疾病(如系统性红斑狼疮)。合理控制I型干扰素的表达具有重要的生物学意义。
I型感染素的产生始于免疫细胞表面的模式识别受体(PRRs)对病毒等的病原体相关分子模式(PAMPs)的识别,通过TLR3/4-TRIF、RIG-I-MAVS和cGAS-STING信号转导通路触发核心转录因子IRF3的磷酸化,进而驱动I型干扰素的转录。值得关注的是,在高剂量的RNA病毒感染情况下,磷酸化IRF3蛋白(p-IRF3)在驱动I型干扰素转录之后,发生完全降解。这个现象表明机体通过促进p-IRF3降解从而避免过量IFNs产生以及不可控的炎症反应发生。然而,目前关于介导p-IRF3完全降解的关键机制并不清楚。研究人员首先通过质谱检测发现在细胞受到病毒刺激后,E3泛素连接酶RAD18与IRF3的结合大大增加,且能促进p-IRF3发生强烈的泛素化,而该泛素化与IRF3的自噬途径降解相关。进一步的研究发现RAD18只结合二聚体 p-IRF3,且二聚体 p-IRF3与DNA链的结合是其被RAD18蛋白识别的前置条件,这提示RAD18可能参与p-IRF3转录活性的调控。当细胞RAD18敲除后,p-IRF3蛋白的降解被抑制且滞留在细胞核内,同时细胞IFNB1的mRNA持续高表达,对病毒(包括SARS-CoV-2)的感染表现出更强的防御反应。机制研究发现E3泛素连接酶RAD18通过其独特的分子识别机制,特异性结合IFNB1启动子上的磷酸化IRF3,并催化形成K63连接的多泛素链修饰(靶向人IRF3的Lys193位点,对应小鼠IRF3的Lys188位点),这种泛素化修饰导致p-IRF3二聚体从转录复合物中解离,并触发其核质转运过程,从而终止IFNB1的转录激活。而其他与IRF3相关E3泛素连接酶(包括TRIM21、TRIM26、RBCK1和UBE3C等)均不具备直接调控p-IRF3转录活性的功能,突显了RAD18在该通路中的特异性调控地位。
研究人员通过Knock-in技术构建了IRF3-K188R(赖氨酸突变为精氨酸)基因突变小鼠,实验结果显示突变小鼠的巨噬细胞在应对病毒感染后,表现出更强的I型IFN反应。实验性变态反应性脑脊髓炎(EAE)造模结果也显示,突变小鼠因产生了更多的IFNβ,其脊髓炎症细胞浸润程度和脊髓脱髓鞘程度均较轻。研究人员还构建了Rad18fl/fl Lysm-cre 小鼠,体内病毒感染实验结果表明Rad18fl/fl Lysm-cre 小鼠在感染水泡性口炎病毒后的生存期更久,体内脏器和血清中的IFNβ因子表达更高。此外,研究人员的临床病理样本分析发现RAD18在系统性红斑狼疮(SLE)病人的外周血单核细胞中的表达较低,在活动期SLE患者中,RAD18蛋白水平分别与IRF3磷酸化水平和IFNB1/IFNA mRNA水平呈负相关,表明RAD18在SLE中对p-IRF3驱动的IFN表达具有负调控作用。这些研究内容为RAD18作为自身免疫性疾病的潜在治疗靶点提供了依据。
图 RAD18调控p-IRF3转录活性的工作模式图
该研究详细阐明RAD18作为E3泛素连接酶在p-IRF3自噬降解通路中的核心调控作用。其分子机制表现为:RAD18特异性识别并结合IFNB启动子区域的p-IRF3二聚体,通过泛素化修饰终止p-IRF3的转录活性,从而精准调控IFNB1基因的表达。这一发现揭示了RAD18在IRF3信号通路中作为“制动器”的关键功能。
浙江大学医学院基础医学院的陈玮和浙大二院眼科中心的韩伟教授为该论文的通信作者。浙江大学医学院博士生蔡依廷、郑家琦博士、赵琳琳硕士和王雪硕士为本文共同第一作者。该研究得到国家自然科学基金和浙江省自然科学基金资助,并得到浙江大学免疫学研究所、免疫与炎症全国重点实验室和浙江大学医学院公共技术平台的帮助和支持。
原文链接:https://www.nature.com/articles/s41590-025-02256-x