
The Immune Landscape and Dynamics of Liver Cancer Revealed by Single-cell RNA Sequencing
Source:Zemin Zhang
2019-11-13
Hepatocellular carcinoma is one of the top three cancers in terms of mortality, with China being one of the worst hit. Immune escape is one of the key factors leading to the development of cancer. Studying the status and dynamic characteristics of different immune cells in tumor microenvironment and further illustrating the potential immune escape mechanisms is of great significance for understanding cancer and controlling and curing cancer. The rapid development of single cell sequencing technology in recent years provides a powerful means to study the status and dynamics of different types of immune cells.
On October 31st, 2019, Zemin Zhang and Xianwen Ren from Biomedical Pioneering Innovation Center (BIOPIC) of Peking University, together with Jirun Peng from Beijing Shijitan Hospital affiliated to the Capital University of Medical Sciences and Kang Liu from Boehringer Ingelheim published a research paper in Cell, entitled “Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma”.
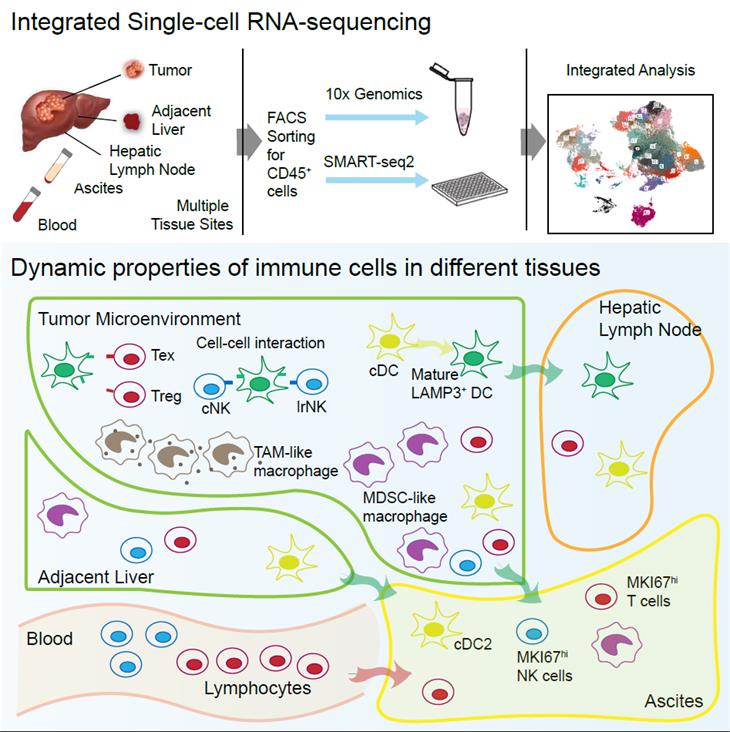
The paper combines 10x Genomics and SMART-seq2, two single-cell sequencing techniques with different advantages in cell and gene capture efficiency, and completed the sequencing and bioinformatics analysis of CD45+ immune cells from liver cancer tissues, tumor-adjacent tissues, lymph nodes, peripheral blood and ascites of 16 liver cancer patients, providing a landscape with single-cell resolution of the tumor immune microenvironment for liver cancers. By integrating transcriptome similarity analysis, RNA velocity analysis, and mitochondrial mutation analysis, the researchers found that immune cells in ascites may have different tissue sources, among which myeloid cells mainly come from tumor and tumor-adjacent tissues while lymphocytes mainly come from blood. Further analysis showed that different groups of immune cells in the tumor had different potential to egress into ascites, among which effector memory T cells and proliferating T cells were more likely to transfer to ascites, while exhausted CD8+ T cells and regulatory T cells were more likely to stay in the tumor. Similar to T cells, different subpopulations of macrophages have different transcriptional states and the ability to transfer to ascites. In the case of dendritic cells, the researchers identified a group of dendritic cells that had high LAMP3 gene expression. This group of LAMP3+ dendritic cells is characterized by activation and maturation, and can migrate from tumors to hepatic lymph nodes, expressing multiple immune-relevant ligand genes and thus interacting with multiple T lymphocyte types. By constructing experimental systems in vitro, the researchers recapitulated these biological characteristics of LAMP3+ dendritic cells observed in vivo, and revealed with The Cancer Genome Atlas data that tumour abundance LAMP3+ dendritic cells is positively associated with the infiltration levels of exhausted CD8+ T cells and regulatory T cells. Therefore, the comprehensive use of single-cell transcriptome sequencing technology with different advantages to analyze the tumor microenvironment may reveal more mechanisms of tumor immune escape, and represents a new research paradigm of related topics.
Qiming Zhang from BIOPIC and College of Life Sciences of Peking University, Yao He from Academy for Advanced Interdisciplinary Studies of Peking University, and Nan Luo from the Ninth School of Clinical Medicine of Peking University 9 clinical medical college of Peking University (Beijing Shijitan Hospital) are the first authors of the paper. Zemin Zhang from BIOPIC, College of Life Sciences, Academy for Advanced Interdisciplinary Studies, and Peking-Tsinghua Center for Life Sciences of Peking university, Xianwen Ren from BIOPIC and College of Life Sciences of Peking University, Jirun Peng from Beijing Shijitan Hospital affiliated to the Capital University of Medical Sciences, and Kang Liu from Boehringer Ingelheim are the corresponding authors of the paper. The project was funded by the Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics with the assistance and support from the high-throughput sequencing platform of Peking University.
On October 31st, 2019, Zemin Zhang and Xianwen Ren from Biomedical Pioneering Innovation Center (BIOPIC) of Peking University, together with Jirun Peng from Beijing Shijitan Hospital affiliated to the Capital University of Medical Sciences and Kang Liu from Boehringer Ingelheim published a research paper in Cell, entitled “Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma”.
The paper combines 10x Genomics and SMART-seq2, two single-cell sequencing techniques with different advantages in cell and gene capture efficiency, and completed the sequencing and bioinformatics analysis of CD45+ immune cells from liver cancer tissues, tumor-adjacent tissues, lymph nodes, peripheral blood and ascites of 16 liver cancer patients, providing a landscape with single-cell resolution of the tumor immune microenvironment for liver cancers. By integrating transcriptome similarity analysis, RNA velocity analysis, and mitochondrial mutation analysis, the researchers found that immune cells in ascites may have different tissue sources, among which myeloid cells mainly come from tumor and tumor-adjacent tissues while lymphocytes mainly come from blood. Further analysis showed that different groups of immune cells in the tumor had different potential to egress into ascites, among which effector memory T cells and proliferating T cells were more likely to transfer to ascites, while exhausted CD8+ T cells and regulatory T cells were more likely to stay in the tumor. Similar to T cells, different subpopulations of macrophages have different transcriptional states and the ability to transfer to ascites. In the case of dendritic cells, the researchers identified a group of dendritic cells that had high LAMP3 gene expression. This group of LAMP3+ dendritic cells is characterized by activation and maturation, and can migrate from tumors to hepatic lymph nodes, expressing multiple immune-relevant ligand genes and thus interacting with multiple T lymphocyte types. By constructing experimental systems in vitro, the researchers recapitulated these biological characteristics of LAMP3+ dendritic cells observed in vivo, and revealed with The Cancer Genome Atlas data that tumour abundance LAMP3+ dendritic cells is positively associated with the infiltration levels of exhausted CD8+ T cells and regulatory T cells. Therefore, the comprehensive use of single-cell transcriptome sequencing technology with different advantages to analyze the tumor microenvironment may reveal more mechanisms of tumor immune escape, and represents a new research paradigm of related topics.
Qiming Zhang from BIOPIC and College of Life Sciences of Peking University, Yao He from Academy for Advanced Interdisciplinary Studies of Peking University, and Nan Luo from the Ninth School of Clinical Medicine of Peking University 9 clinical medical college of Peking University (Beijing Shijitan Hospital) are the first authors of the paper. Zemin Zhang from BIOPIC, College of Life Sciences, Academy for Advanced Interdisciplinary Studies, and Peking-Tsinghua Center for Life Sciences of Peking university, Xianwen Ren from BIOPIC and College of Life Sciences of Peking University, Jirun Peng from Beijing Shijitan Hospital affiliated to the Capital University of Medical Sciences, and Kang Liu from Boehringer Ingelheim are the corresponding authors of the paper. The project was funded by the Natural Science Foundation of China and the Beijing Advanced Innovation Center for Genomics with the assistance and support from the high-throughput sequencing platform of Peking University.
