
Xuan Zhang's team identified novel mechanism of systemic lupus erythematosus
Source:Xuan Zhang
2021-09-16
Systemic lupus erythematosus (SLE) is a chronic debilitating autoimmune disease characterized by a global loss of immune tolerance with activation of both innate and adaptive branches of the immune system. Central features include accumulated non-cleared cell debris derived from various forms of cell death, elevated type I IFN signaling and increased autoantibody production. Neutrophils are the dominant immune cells in the circulation and contribute to a variety of autoimmune disorders. In patients with SLE, a number of abnormalities in neutrophils have been reported, including impaired phagocytosis, increased aggregation and accelerated cell death. Elevated levels of anti-neutrophil cytoplasmic antibodies in the sera of patients with SLE suggest that neutrophil death together with ineffective clearance of the subsequent debris provide a stable source of autoantigens for disease initiation and propagation. However, the main forms of neutrophil death in SLE and the underlying mechanisms have not been fully characterized.
On August 12, 2021, Professor Xuan Zhang from the Clinical Immunology Center, Peking Union Medical College, Chinese Academy of Medical Sciences/Beijing Hospital led the group with collaborators from Harvard Medical School to publish a paper in Nature Immunology titled " Glutathione peroxidase 4 regulated neutrophil ferroptosis induces systemic autoimmunity ". For the first time, the key role of neutrophil ferroptosis in autoimmune diseases in both patients and animal models as well as the underlying regulatory mechanisms of the specific neutrophil death in SLE patients are identified and explored. The study identifies the critical role of intrinsic immune cell abnormalities in systemic autoimmunity, overturning the traditional conception that SLE is primarily caused by abnormalities in adaptive immunity.
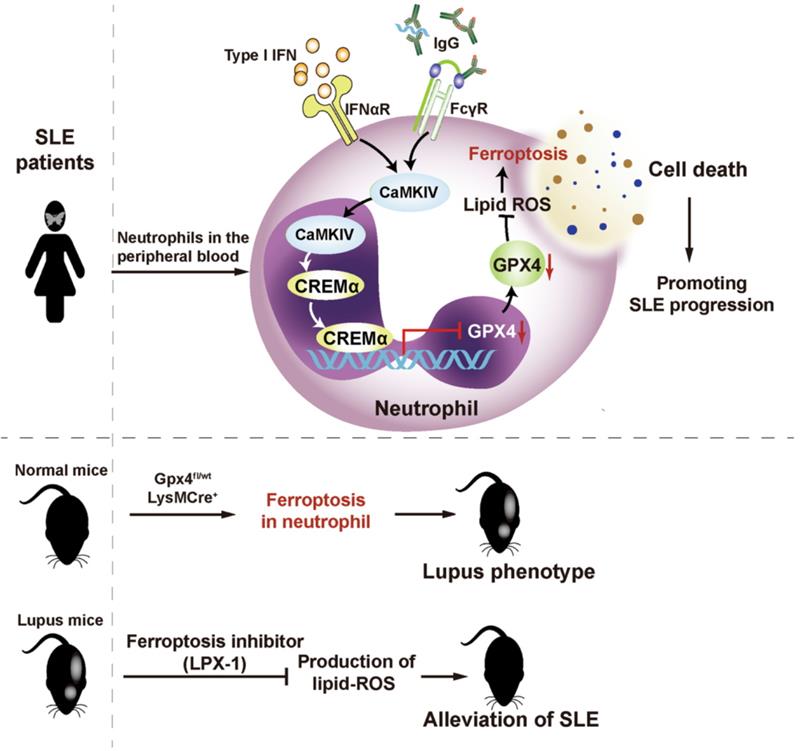
Ferroptosis is a newly recognized form of programmed cell death that is morphologically, biochemically, and genetically distinct from other forms of regulated cell death such as apoptosis, necroptosis and pyroptosis. It is characterized by production of lipid-reactive oxygen species (ROS) and iron overload, leading to caspase- and necrosome-independent cell death. As a key regulator of ferroptosis, phospholipid peroxidase glutathione peroxidase 4 (GPX4) can detoxify hydroperoxides in membrane lipids directly, thereby reducing damage to membrane function and preventing the generation of lipid peroxidation-derived reactive products and mitigating ferroptosis. Inhibition or ablation of GPX4 induces ferroptosis in different cell types.
First, the researchers found that patients with SLE exhibited significantly lower neutrophil counts than healthy individuals and patients with other immune diseases, as well as significantly lower cell viability. The above-mentioned phenomena could be alleviated after clinical treatment. The team hypothesized that the phenomenon was serum-related and examined serum inflammatory factor profiles in SLE patients, and revealed that elevated autoantibodies and type I interferon in SLE could significantly induce neutrophil death. To investigate the specific form of neutrophil death, the research team examined the morphology of SLE neutrophils by electron microscopy and found that their mitochondria showed characteristics of ferroptosis. Meanwhile, in vitro experiments demonstrated that inhibition of ferroptosis significantly alleviated lupus serum-induced neutrophil death. In vivo experiments in mice further confirmed that inhibition of ferroptosis could mitigate the progression of SLE. Based on these findings, the team constructed a neutrophil ferroptosis-sensitive mouse model with gene editing technology, which exhibited skin lesions, lymph node enlargement, elevated urine proteins, and presence of autoantibodies, successfully mimicking the symptoms of SLE.
By transcriptome sequencing of neutrophils, the team found that 23 ferroptosis-related genes were expressed abnormally in SLE patients. Therein, GPX4 significantly reduced and the degree of reduction correlated with SLE disease activity. To investigate the specific mechanisms underlying the reduction of GPX4 and consequent ferroptosis in SLE neutrophils, the researchers employed chromatin immunoprecipitation and DNA-pulldown, finding that the promoter region of GPX4 in neutrophils from patients bound to the CREM-repressed transcription factor abnormally. And SLE serum, type I interferon or SLE IgG, could activate the CaMKIV, further promoting the binding of CREM to the promoter region of GPX4, thereby inhibiting GPX4 transcription.
Prof. Zhang said, "Although most previous studies focused on the role of adaptive immunity in triggering systemic autoimmune diseases, our study has somehow overturned the traditional concept by revealing that abnormalities in innate immune cells alone can lead to the development of systemic autoimmunity. The role of ferroptosis in autoimmune diseases has not been reported previously, and our study finds that it is not only an important cause of leukopenia in SLE patients, but also induces the occurrence of cascade pathological reactions and in turn promote the development of SLE."
The study, based on a SLE cohort covering Asian, Caucasian and African American populations, confirmed the critical role of intrinsic immune cell abnormalities in systemic autoimmunity by in vivo and in vitro experiments, knockout gene models and transcriptomics, providing a potential new treatment target for SLE.
The article was co-first authored by Pengchong Li, Mengdi Jiang, Ketian Li, from Peking Union Medical College and Hao Li from Harvard Medical School.
Links: https://www.nature.com/articles/s41590-021-00993-3
On August 12, 2021, Professor Xuan Zhang from the Clinical Immunology Center, Peking Union Medical College, Chinese Academy of Medical Sciences/Beijing Hospital led the group with collaborators from Harvard Medical School to publish a paper in Nature Immunology titled " Glutathione peroxidase 4 regulated neutrophil ferroptosis induces systemic autoimmunity ". For the first time, the key role of neutrophil ferroptosis in autoimmune diseases in both patients and animal models as well as the underlying regulatory mechanisms of the specific neutrophil death in SLE patients are identified and explored. The study identifies the critical role of intrinsic immune cell abnormalities in systemic autoimmunity, overturning the traditional conception that SLE is primarily caused by abnormalities in adaptive immunity.
Ferroptosis is a newly recognized form of programmed cell death that is morphologically, biochemically, and genetically distinct from other forms of regulated cell death such as apoptosis, necroptosis and pyroptosis. It is characterized by production of lipid-reactive oxygen species (ROS) and iron overload, leading to caspase- and necrosome-independent cell death. As a key regulator of ferroptosis, phospholipid peroxidase glutathione peroxidase 4 (GPX4) can detoxify hydroperoxides in membrane lipids directly, thereby reducing damage to membrane function and preventing the generation of lipid peroxidation-derived reactive products and mitigating ferroptosis. Inhibition or ablation of GPX4 induces ferroptosis in different cell types.
First, the researchers found that patients with SLE exhibited significantly lower neutrophil counts than healthy individuals and patients with other immune diseases, as well as significantly lower cell viability. The above-mentioned phenomena could be alleviated after clinical treatment. The team hypothesized that the phenomenon was serum-related and examined serum inflammatory factor profiles in SLE patients, and revealed that elevated autoantibodies and type I interferon in SLE could significantly induce neutrophil death. To investigate the specific form of neutrophil death, the research team examined the morphology of SLE neutrophils by electron microscopy and found that their mitochondria showed characteristics of ferroptosis. Meanwhile, in vitro experiments demonstrated that inhibition of ferroptosis significantly alleviated lupus serum-induced neutrophil death. In vivo experiments in mice further confirmed that inhibition of ferroptosis could mitigate the progression of SLE. Based on these findings, the team constructed a neutrophil ferroptosis-sensitive mouse model with gene editing technology, which exhibited skin lesions, lymph node enlargement, elevated urine proteins, and presence of autoantibodies, successfully mimicking the symptoms of SLE.
By transcriptome sequencing of neutrophils, the team found that 23 ferroptosis-related genes were expressed abnormally in SLE patients. Therein, GPX4 significantly reduced and the degree of reduction correlated with SLE disease activity. To investigate the specific mechanisms underlying the reduction of GPX4 and consequent ferroptosis in SLE neutrophils, the researchers employed chromatin immunoprecipitation and DNA-pulldown, finding that the promoter region of GPX4 in neutrophils from patients bound to the CREM-repressed transcription factor abnormally. And SLE serum, type I interferon or SLE IgG, could activate the CaMKIV, further promoting the binding of CREM to the promoter region of GPX4, thereby inhibiting GPX4 transcription.
Figure: The role of neutrophil iron death in the pathogenesis of SLE. (Image source: Nat Immunol)
Prof. Zhang said, "Although most previous studies focused on the role of adaptive immunity in triggering systemic autoimmune diseases, our study has somehow overturned the traditional concept by revealing that abnormalities in innate immune cells alone can lead to the development of systemic autoimmunity. The role of ferroptosis in autoimmune diseases has not been reported previously, and our study finds that it is not only an important cause of leukopenia in SLE patients, but also induces the occurrence of cascade pathological reactions and in turn promote the development of SLE."
The study, based on a SLE cohort covering Asian, Caucasian and African American populations, confirmed the critical role of intrinsic immune cell abnormalities in systemic autoimmunity by in vivo and in vitro experiments, knockout gene models and transcriptomics, providing a potential new treatment target for SLE.
The article was co-first authored by Pengchong Li, Mengdi Jiang, Ketian Li, from Peking Union Medical College and Hao Li from Harvard Medical School.
Links: https://www.nature.com/articles/s41590-021-00993-3
