
Dr. Qiming Liang’s team found new activation mechanism of NLRP1 inflammasome
Source:Qiming Liang
2022-07-06
Inflammasomes are large multiprotein complexes that sense and amplify signals from various stimuli to activate inflammatory caspases, leading to the production of inflammatory cytokines and pyroptotic cell death. NLRP1 is the first protein in the NOD-like receptor family found to form a complex with ASC and caspase1. Unlike other NLRP proteins, hNLRP1 has a distinct C-terminal region containing a function-to-find domain (FIIND) and a CARD2. hNLRP1 is the only NLR that undergoes constitutive post-translational autoproteolysis at position Ser1213 between the subdomains ZU5 and UPA in the FIIND, generating two polypeptides N-terminal NLRP1 (NLRP1NT) and C-terminal NLRP1 (NLRP1CT), which remain associated with each other. The cleavage in FIIND is not fully processed, and only a fraction of the total NLRP1 protein undergoes autoproteolysis. NLRP1CT can form an autoinhibtion complex with NLRP1NT and uncleaved full-length NLRP1FL to inhibit its own activity.
The current understanding of NLRP1 inflammasome activation is largely limited to NLRP1NT degradation by pathogen-encoded proteases. The Bacillus anthracis lethal toxin cleaves murine NLRP1b between amino acid positions 44 and 45, resulting in the proteasomal degradation of NLRP1bNT. The enterovirus 3C protease (3CPro) directly cleaves hNLRP1 between amino acids 130 and 131 in the Linker1 region, triggering the degradation of hNLRP1NT and the activation of the hNLRP1 inflammasome. The host protease DPP9 forms a ternary complex with full-length NLRP1 (NLRP1FL) and NLRP1CT to sequester NLRP1CT and prevent its oligomerization. Val-boroPro (VbP), a DPP8–DPP9 inhibitor, weakens the hNLRP1–DPP9 interaction and accelerates the degradation of hNLRP1NT, leading to inflammasome activation.
Dr. Liang’s group found that KSHV infection can induce the assembly and activation of the inflammasome. The activation depends on activation by a viral protein called ORF45 in the KSHV rather than the recognition of viral DNA or cellular DNA receptor IFI16. The research group identified human NLRP1 as a receptor for ORF45 to activate the inflammasome through yeast two-hybrid screening. Knockout of NLRP1 in cells inhibited the inflammasome activation induced by KSHV infection or ORF45 expression. Knockout of ORF45 in KSHV (KSHV△ORF45) also failed to induce inflammasome activation. The Linker1 region located between the PYD and NACHT domains is crucial for the formation of human NLRP1 autoinhibition, and Linker1 binds to the UPA domain of NLRP1CT to limit the activation of NLRP1CT. ORF45 competitively inhibits the interaction between NLRP1NT and NLRP1CT by binding to Linker1, so that the activated NLRP1CT can assemble with ASC and caspase1 to form an inflammasome in the cytoplasm. Moreover, NLRP1NT can translocate into the nucleus without classical functional degradation (Figure). Meanwhile, activation of NLRP1 by ORF45 is well conserved in primates. ORF45 does not affect the binding between NLRP1 and DPP9. VbP does not affect the activation of NLRP1 by ORF45.
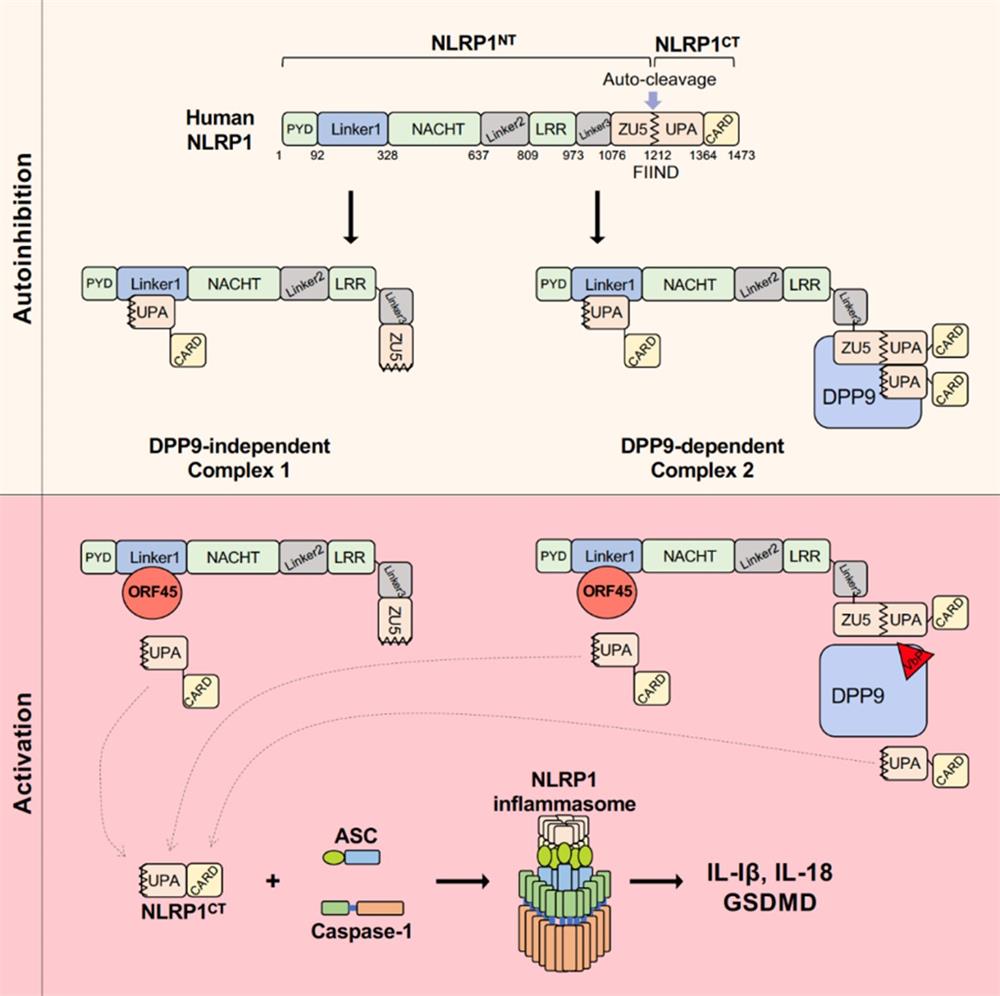
This study reveals a novel non-canonical activation pathway of NLRP1 independent of NLRP1NT degradation, shedding light for the regulatory mechanism of NLRP1 auto-inhibition and activation. This study elucidates that KSHV structural protein rather than viral DNA is the main ligand for NLRP1 inflammasome.
This work was supported by grants from the National Key Research and Development Project of China (2018YFA0900802), the National Natural Science Foundation of China (31770176), Shanghai Frontiers Science Center of Cellular Homeostasis and Human Disease, and the Shanghai Municipal Health Commission (201940179). We thank Dr. Feng Shao (National Institute of Biological Sciences, China) for valuable suggestions.
The current understanding of NLRP1 inflammasome activation is largely limited to NLRP1NT degradation by pathogen-encoded proteases. The Bacillus anthracis lethal toxin cleaves murine NLRP1b between amino acid positions 44 and 45, resulting in the proteasomal degradation of NLRP1bNT. The enterovirus 3C protease (3CPro) directly cleaves hNLRP1 between amino acids 130 and 131 in the Linker1 region, triggering the degradation of hNLRP1NT and the activation of the hNLRP1 inflammasome. The host protease DPP9 forms a ternary complex with full-length NLRP1 (NLRP1FL) and NLRP1CT to sequester NLRP1CT and prevent its oligomerization. Val-boroPro (VbP), a DPP8–DPP9 inhibitor, weakens the hNLRP1–DPP9 interaction and accelerates the degradation of hNLRP1NT, leading to inflammasome activation.
Dr. Liang’s group found that KSHV infection can induce the assembly and activation of the inflammasome. The activation depends on activation by a viral protein called ORF45 in the KSHV rather than the recognition of viral DNA or cellular DNA receptor IFI16. The research group identified human NLRP1 as a receptor for ORF45 to activate the inflammasome through yeast two-hybrid screening. Knockout of NLRP1 in cells inhibited the inflammasome activation induced by KSHV infection or ORF45 expression. Knockout of ORF45 in KSHV (KSHV△ORF45) also failed to induce inflammasome activation. The Linker1 region located between the PYD and NACHT domains is crucial for the formation of human NLRP1 autoinhibition, and Linker1 binds to the UPA domain of NLRP1CT to limit the activation of NLRP1CT. ORF45 competitively inhibits the interaction between NLRP1NT and NLRP1CT by binding to Linker1, so that the activated NLRP1CT can assemble with ASC and caspase1 to form an inflammasome in the cytoplasm. Moreover, NLRP1NT can translocate into the nucleus without classical functional degradation (Figure). Meanwhile, activation of NLRP1 by ORF45 is well conserved in primates. ORF45 does not affect the binding between NLRP1 and DPP9. VbP does not affect the activation of NLRP1 by ORF45.
This study reveals a novel non-canonical activation pathway of NLRP1 independent of NLRP1NT degradation, shedding light for the regulatory mechanism of NLRP1 auto-inhibition and activation. This study elucidates that KSHV structural protein rather than viral DNA is the main ligand for NLRP1 inflammasome.
This work was supported by grants from the National Key Research and Development Project of China (2018YFA0900802), the National Natural Science Foundation of China (31770176), Shanghai Frontiers Science Center of Cellular Homeostasis and Human Disease, and the Shanghai Municipal Health Commission (201940179). We thank Dr. Feng Shao (National Institute of Biological Sciences, China) for valuable suggestions.
