
Ning Wu’team discovered lipid asymmetry disruption by XKR8 orchestrates neutrophil extracellular trap formation and inhibits fungal infection
Source:Ning Wu
2026-05-19
On March 4, 2026, the research group led by Professor Ning Wu published a paper titled "Lipid asymmetry disruption by XKR8 orchestrates neutrophil extracellular trap formation and inhibits fungal infection" in Nature Immunology. This study identifies the central role of XKR8-mediated lipid redistribution in the formation of neutrophil extracellular traps (NETs). It reveals a novel mechanism by which XKR8 regulates plasma membrane lipid asymmetry during NET formation while simultaneously activating mechanosensitive calcium channels on the cell membrane, thereby promoting NET release. This work not only establishes XKR8-dependent lipid asymmetry as a critical regulator of NET formation but also provides promising therapeutic targets for fungal infections, acute lung injury, and autoimmune diseases.
Neutrophils, the most abundant white blood cells in human and mammalian peripheral blood, play an essential role in defending against various pathogens, including bacteria and fungi. Their functions, such as phagocytosis, degranulation, and NET formation, help protect the body from infection. Neutrophil extracellular traps were first reported by Brinkmann et al. in 2004, who defined the unique programmed cell death as NETosis. During NETosis, neutrophils decondense their nuclear chromatin and release their DNA along with myeloperoxidase (MPO), neutrophil elastase (NE), and other antimicrobial proteins, forming web-like structures that trap and kill pathogens, thereby preventing their spread.
However, excessive neutrophil activation and uncontrolled NET formation contribute to many diseases, including acute respiratory distress syndrome (ARDS), rheumatoid arthritis (RA), systemic lupus erythematosus, thrombosis, and tumor metastasis. Therefore, elucidating the molecular mechanisms that regulate NET formation can not only enhance the body's ability to defend against pathogen infections but also provide strategies for treating NET-related diseases.
To address this question, the researchers purified neutrophils from human peripheral blood and mouse bone marrow and subsequently stimulated them with the classical NET inducer PMA. Using flow cytometry and microscopy, they observed that plasma membrane (PM) lipid redistribution, monitored by phosphatidylserine (PS) exposure, preceded PM permeabilization and subsequent NET release in both bone marrow-derived and human peripheral blood neutrophils. To identify the responsible scramblase, they generated Tmem16f and Xkr8 germline knockout (KO) mice. The results demonstrated that XKR8-deficient neutrophils exhibited severely impaired PS exposure during PMA-induced NET formation, whereas TMEM16F-deficient neutrophils did not. Remarkably, XKR8 KO neutrophils failed to form NETs in response to classical chemical inducers (e.g., PMA, A23187, LPS) as well as pathogens (e.g., Candida albicans, Listeria monocytogenes, Staphylococcus aureus, and Escherichia coli).
XKR8 was originally identified as a phospholipid scramblase activated by caspase-3 during apoptosis. To determine whether caspase-3 participates in NET formation, the researchers used western blotting and immunofluorescence and confirmed that caspase-3 is activated during PMA-induced NET formation. Moreover, treatment with caspase-3 inhibitors (Q-VD-OPh or emricasan) and the use of Casp3 KO neutrophils resulted in severely impaired PS exposure and NET formation upon PMA stimulation. Furthermore, the researchers generated Xkr8 2DA mutant (caspase-3 cleavage-site mutant) knock-in mice and HL-60 cells overexpressing the 2DA mutant. Neutrophils bearing the XKR8 2DA mutation exhibited a severe defect in PS exposure and NET formation in response to various inducers. Collectively, these findings indicate that activated caspase-3 cleaves XKR8, which in turn triggers disruption of lipid asymmetry.
The previous results showed that XKR8 KO neutrophils failed to generate citrullinated histone H3 (Cit-H3), suggesting impaired PAD4 activation. The researchers therefore hypothesized that calcium signaling mediated by lipid scrambling participates in NET formation. They found that treatment with the calcium chelators EGTA or BAPTA-AM at the onset of PMA stimulation completely blocked NET formation. However, when the chelators were added three hours after stimulation, by which time PS externalization had already occurred, NET formation was not abolished. This suggests that PS exposure contributes to the initial intracellular calcium accumulation during NET formation. To further test this hypothesis, a membrane tension sensitive probe, Flipper-TR, was used to monitor membrane tension via fluorescence lifetime imaging microscopy (FLIM). Notably, the fluorescence lifetime of Flipper-TR was significantly reduced in living neutrophils undergoing lipid scrambling, indicating a marked decrease in membrane tension. Using small-molecule inhibitors and CRISPR-Cas9 technology, they identified that mechanosensitive calcium channels on the plasma membrane, including TRPV2, TRPC3, Piezo1, and TRPV4, facilitate NET formation. Together, these results reveal a novel regulatory axis in NET formation: the caspase-3-XKR8-calcium signaling axis.
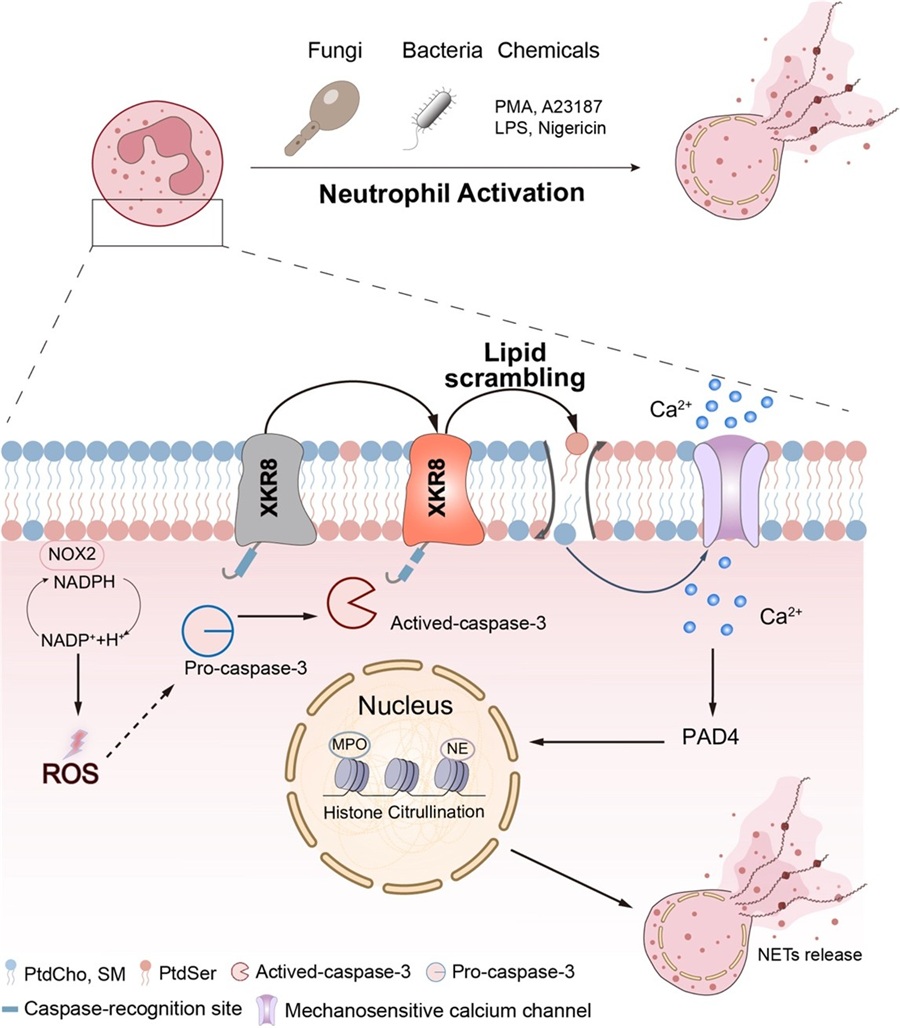
To assess the in vivo role of XKR8 in NET formation, the researchers established acute lung injury (ALI) models, collagen antibody–induced arthritis (CAIA) models, and Candida albicans infection models using XKR8 KO mice, neutrophil-specific XKR8 KO mice, and XKR8 2DA mutant knock-in mice. The results showed that XKR8 deficiency led to significantly reduced inflammation in both ALI and CAIA models, likely due to decreased NET formation. In contrast, loss of XKR8 in neutrophils exacerbated fungal infection. Importantly, treatment with the TRPV4 agonist GSK1016790A bypassed the XKR8 deficiency and restored antifungal immunity, offering a novel therapeutic strategy for fungal infection.
Professor Ning Wu (The First Affiliated Hospital of Anhui Medical University and Institute of Clinical Immunology, Anhui Medical University; Department of Immunology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology), Professor Zheng Liu (Department of Otorhinolaryngology, Head and Neck Surgery, Zhongnan Hospital of Wuhan University), and Professor Wei Hu (Department of Clinical Pharmacology, The Second Affiliated Hospital of Anhui Medical University) are the corresponding authors. Weixiang Liu, Jieming Ping (PhD students at The First Affiliated Hospital of Anhui Medical University and Institute of Clinical Immunology, Anhui Medical University; Department of Immunology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology), and Lishan Deng (Master student at the Department of Otolaryngology–Head and Neck Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology) are the co-first authors.
Article link: https://www.nature.com/articles/s41590-026-02456-z
Neutrophils, the most abundant white blood cells in human and mammalian peripheral blood, play an essential role in defending against various pathogens, including bacteria and fungi. Their functions, such as phagocytosis, degranulation, and NET formation, help protect the body from infection. Neutrophil extracellular traps were first reported by Brinkmann et al. in 2004, who defined the unique programmed cell death as NETosis. During NETosis, neutrophils decondense their nuclear chromatin and release their DNA along with myeloperoxidase (MPO), neutrophil elastase (NE), and other antimicrobial proteins, forming web-like structures that trap and kill pathogens, thereby preventing their spread.
However, excessive neutrophil activation and uncontrolled NET formation contribute to many diseases, including acute respiratory distress syndrome (ARDS), rheumatoid arthritis (RA), systemic lupus erythematosus, thrombosis, and tumor metastasis. Therefore, elucidating the molecular mechanisms that regulate NET formation can not only enhance the body's ability to defend against pathogen infections but also provide strategies for treating NET-related diseases.
To address this question, the researchers purified neutrophils from human peripheral blood and mouse bone marrow and subsequently stimulated them with the classical NET inducer PMA. Using flow cytometry and microscopy, they observed that plasma membrane (PM) lipid redistribution, monitored by phosphatidylserine (PS) exposure, preceded PM permeabilization and subsequent NET release in both bone marrow-derived and human peripheral blood neutrophils. To identify the responsible scramblase, they generated Tmem16f and Xkr8 germline knockout (KO) mice. The results demonstrated that XKR8-deficient neutrophils exhibited severely impaired PS exposure during PMA-induced NET formation, whereas TMEM16F-deficient neutrophils did not. Remarkably, XKR8 KO neutrophils failed to form NETs in response to classical chemical inducers (e.g., PMA, A23187, LPS) as well as pathogens (e.g., Candida albicans, Listeria monocytogenes, Staphylococcus aureus, and Escherichia coli).
XKR8 was originally identified as a phospholipid scramblase activated by caspase-3 during apoptosis. To determine whether caspase-3 participates in NET formation, the researchers used western blotting and immunofluorescence and confirmed that caspase-3 is activated during PMA-induced NET formation. Moreover, treatment with caspase-3 inhibitors (Q-VD-OPh or emricasan) and the use of Casp3 KO neutrophils resulted in severely impaired PS exposure and NET formation upon PMA stimulation. Furthermore, the researchers generated Xkr8 2DA mutant (caspase-3 cleavage-site mutant) knock-in mice and HL-60 cells overexpressing the 2DA mutant. Neutrophils bearing the XKR8 2DA mutation exhibited a severe defect in PS exposure and NET formation in response to various inducers. Collectively, these findings indicate that activated caspase-3 cleaves XKR8, which in turn triggers disruption of lipid asymmetry.
The previous results showed that XKR8 KO neutrophils failed to generate citrullinated histone H3 (Cit-H3), suggesting impaired PAD4 activation. The researchers therefore hypothesized that calcium signaling mediated by lipid scrambling participates in NET formation. They found that treatment with the calcium chelators EGTA or BAPTA-AM at the onset of PMA stimulation completely blocked NET formation. However, when the chelators were added three hours after stimulation, by which time PS externalization had already occurred, NET formation was not abolished. This suggests that PS exposure contributes to the initial intracellular calcium accumulation during NET formation. To further test this hypothesis, a membrane tension sensitive probe, Flipper-TR, was used to monitor membrane tension via fluorescence lifetime imaging microscopy (FLIM). Notably, the fluorescence lifetime of Flipper-TR was significantly reduced in living neutrophils undergoing lipid scrambling, indicating a marked decrease in membrane tension. Using small-molecule inhibitors and CRISPR-Cas9 technology, they identified that mechanosensitive calcium channels on the plasma membrane, including TRPV2, TRPC3, Piezo1, and TRPV4, facilitate NET formation. Together, these results reveal a novel regulatory axis in NET formation: the caspase-3-XKR8-calcium signaling axis.
To assess the in vivo role of XKR8 in NET formation, the researchers established acute lung injury (ALI) models, collagen antibody–induced arthritis (CAIA) models, and Candida albicans infection models using XKR8 KO mice, neutrophil-specific XKR8 KO mice, and XKR8 2DA mutant knock-in mice. The results showed that XKR8 deficiency led to significantly reduced inflammation in both ALI and CAIA models, likely due to decreased NET formation. In contrast, loss of XKR8 in neutrophils exacerbated fungal infection. Importantly, treatment with the TRPV4 agonist GSK1016790A bypassed the XKR8 deficiency and restored antifungal immunity, offering a novel therapeutic strategy for fungal infection.
Professor Ning Wu (The First Affiliated Hospital of Anhui Medical University and Institute of Clinical Immunology, Anhui Medical University; Department of Immunology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology), Professor Zheng Liu (Department of Otorhinolaryngology, Head and Neck Surgery, Zhongnan Hospital of Wuhan University), and Professor Wei Hu (Department of Clinical Pharmacology, The Second Affiliated Hospital of Anhui Medical University) are the corresponding authors. Weixiang Liu, Jieming Ping (PhD students at The First Affiliated Hospital of Anhui Medical University and Institute of Clinical Immunology, Anhui Medical University; Department of Immunology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology), and Lishan Deng (Master student at the Department of Otolaryngology–Head and Neck Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology) are the co-first authors.
Article link: https://www.nature.com/articles/s41590-026-02456-z
